

Pesquisadores da Esalq descobrem novas facetas dos vírus gigantes
Métodos incomuns permitiram identificação de pelo menos quatro novas famílias. Organização do grupo é fundamental para entender impacto no ambiente e saúde


{kind=link}
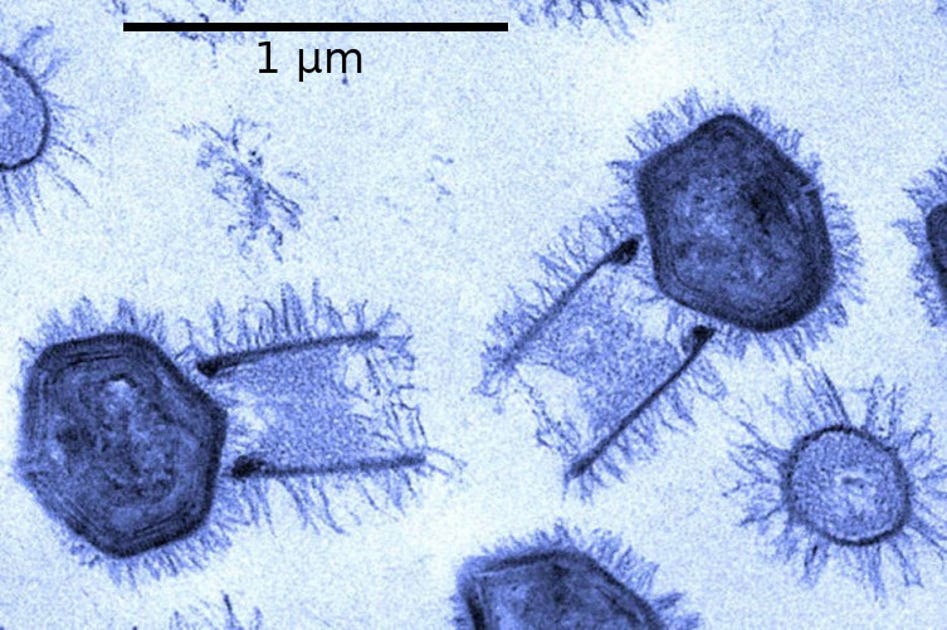
Os vírus gigantes ainda são uma novidade para a virologia. Descritos pela primeira vez nos anos 2000, sua identificação mudou o estudo da virosfera ao revelar uma comunidade biológica até então desconhecida. Na Escola Superior de Agricultura Luiz de Queiroz (Esalq) da USP, pesquisadores expuseram mais uma faceta desconhecida desses organismos.
Um estudo liderado por pesquisadores da Esalq e da Universidade Federal de Minas Gerais (UFMG) propõe uma nova divisão dentro da família de vírus gigantes Asfarviridae– apenas uma das que dividem o mundo dos gigavírus. Ainda que compartilhem uma origem comum, os dados genéticos indicam que os indivíduos hoje pertencentes a um único clado são muito diferentes entre si – eles extrapolam os limites esperados para um grupo.
“Existem pelo menos cinco famílias diferentes dentro do que se imaginava que seria uma”, diz Luiz Eduardo Del Bem, autor correspondente da pesquisa. Professor do Departamento de Genética do instituto, Del Bem afirma que a grande distância evolutiva é evidência de que as espécies devem ser divididas em novas famílias. Vírus gigantes são, na verdade, compostos de “mais espécies e gêneros diferentes do que se imaginava”, segundo o pesquisador.

Luiz-Eduardo Del-Bem – Foto: Reprodução/LinkedIn
Os resultados foram publicados na revista científica Journal of Virology. No periódico, o texto foi agraciado com duas distinções: escolha do editor, dada para cerca de 10% dos artigos, e “gema preciosa”, dada para 1% do que é publicado na revista – apenas textos com o potencial de mudar o paradigma da virologia.

{kind=link}
O que são?
A descoberta dos vírus forçou a biologia a repensar suas categorias e questionar, ainda, o que é um ser vivo. Eram entendidos como entidades simples, parasitas obrigatórios de tamanho sempre muito reduzido – alguns apresentam apenas três ou quatro genes. Esse modelo só começou a mudar com a descoberta das primeiras linhagens gigantes.
“Estão fora da curva”, diz Thiago Mendonça-Santos, primeiro autor do artigo, sobre os vírus gigantes. Ele explica que, de tão grandes, os microrganismos, por vezes, são confundidos com bactérias – algumas ordens de grandeza maiores que vírus comuns.
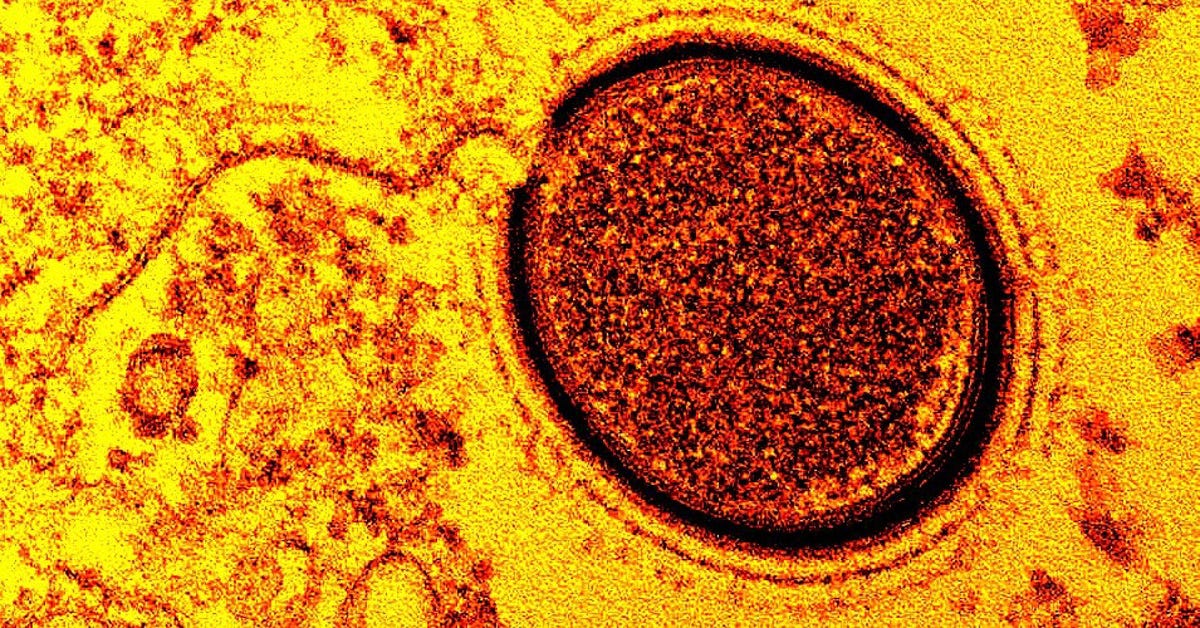
O tamanho espanta, mas a complexidade genética também. Um vírus gigante recém-descoberto chega a 2.600 nanômetros e apresenta 867 genes – metade sem função conhecida pela ciência. O coronavírus, por exemplo, tem 120 e 11, respectivamente.
O trabalho, produto do doutorado de Mendonça-Santos, aponta uma diversidade maior do que antes se imaginava. A descoberta sugere que ainda há muito o que se explorar. Apesar dos avanços, “ainda não se sabe exatamente o que eles são nem quais funções biológicas são capazes de realizar”, afirma Del Bem.
Quebra-cabeças
A descoberta desses microrganismos foi tardia. Por muito tempo, não se sabia que eles sequer existiam. Filtros antes utilizados para a captação de vírus detinham os gigantes junto de bactérias e outros microrganismos unicelulares maiores, que não iam para análise. Mendonça-Santos também explica que as espécies do grupo ainda não são de interesse para a saúde pública: “São ambientais e não infectam humanos”. Portanto, passaram despercebidos.
Os vírus gigantes estudados, antes pertencentes a uma mesma família, infectam uma ampla gama de organismos – de amebas a porcos, seus hospedeiros mais complexos. Segundo os pesquisadores, a amplitude de possibilidades já era um indicativo da diversidade oculta. À medida que os primeiros resultados foram obtidos, Mendonça-Santos questionou: seriam todos realmente membros da mesma família?
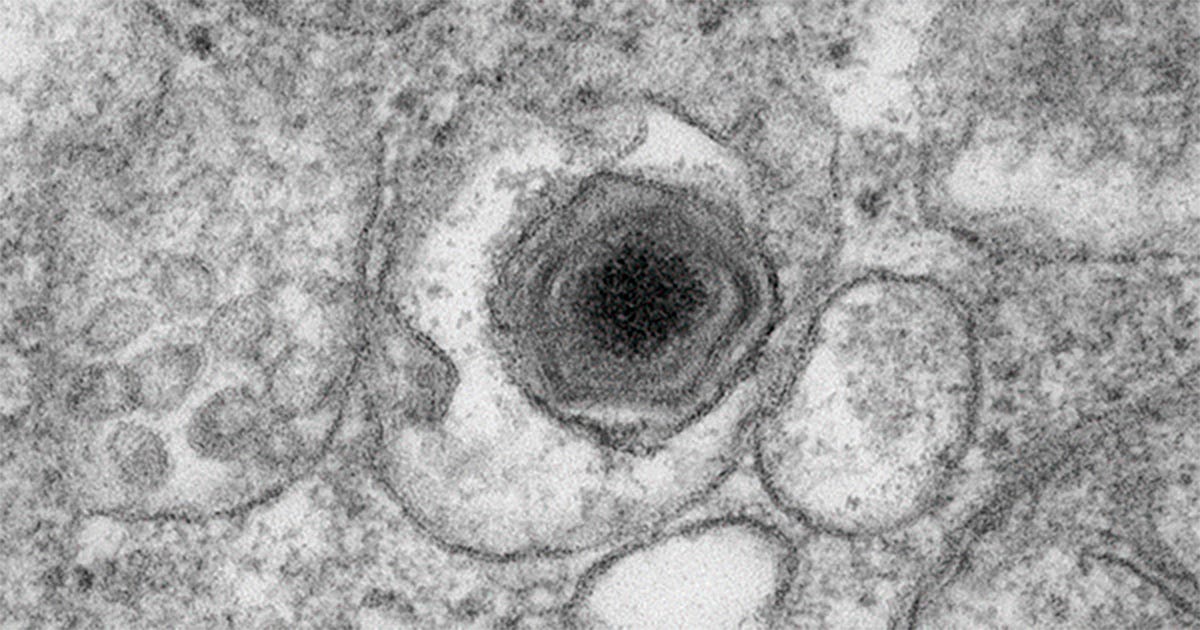
Diante dos 39 genomas completos disponíveis da família Asfarviridae, os pesquisadores constataram a diversidade por meio da aplicação de métodos incomuns: foram “importadas” técnicas genômicas da botânica e do estudo de organismos multicelulares.
Foram identificados 2.483 grupos de genes, dos quais apenas 37 estavam presentes em todos os vírus. Quase 40% dos genes ocorrem apenas em um único genoma, indicando enorme diversidade.
Essas informações foram utilizadas para construir uma nova árvore filogenética. Nela, é possível observar a divisão das linhagens – cada uma formando um agrupamento próprio – e o distanciamento evolutivo entre as espécies –, ou quantos passos são necessários para se chegar ao antepassado comum. “Esses vírus percorreram caminhos distintos e isso precisa aparecer na taxonomia”, explica Del Bem.
Os resultados foram obtidos sem custos. O banco de dados genômicos utilizado é público, fornecido pelo Centro Nacional de Informação Biotecnológica do Instituto Nacional de Saúde (NIH) dos Estados Unidos. A publicação do artigo – condecorado pela revista – também foi gratuita. Diante do sucesso da pesquisa, Del Bem conclui que é possível “fazer uma ciência de alta qualidade com baixíssimo investimento, se as perguntas certas forem feitas”.
{kind=link}
Novas propostas
Mendonça-Santos explica que existe uma entidade que organiza a taxonomia dos vírus, o Comitê Internacional de Taxonomia de Vírus (ICTV). É o órgão que acomoda esses microrganismos nas caixinhas de espécie, gênero, família e ordem – ainda que não sejam considerados seres vivos.
No artigo, os autores propõem uma nova classificação. Manter cada gigavírus antes pertencente à Asfarviridae na mesma família já não fazia sentido – de acordo com os pesquisadores, os dados suportam a reclassificação taxonômica em clados bem definidos.
A família Asfarviridae – agora com apenas três espécies – foi mantida, mas quatro novas foram sugeridas: Faustoviridae, com quatro espécies, Kaumoebaviridae, com duas, Pacmanviridae, com duas, e Abaloneviridae, com um gênero descrito, mas ainda sem espécies identificadas.
A separação foi definida a partir da identidade genética – um índice calculado a partir de dados genômicos – de cada vírus gigante. Quanto mais alto é o valor, mais parecidos e mais próximos são os organismos: acima de 95% de semelhança sugere que sejam da mesma espécie, até 70%, do mesmo gênero, e menos de 40%, da mesma família. “Esses valores [de separação de clados] são arbitrários, mas são baseados em dados experimentais”, afirma Del Bem.
“Ao contrário de outros organismos, em vírus não há tantos critérios morfológicos possíveis de serem usados”, explica Del Bem. A identificação de características genéticas é a solução para classificar as espécies. “É como se tentássemos calcular o fenômeno biológico que estamos vendo através desses critérios.”
O estudo mostra que esses parâmetros foram ultrapassados e destaca que a organização do grupo é necessária para compreender o funcionamento e o impacto dos vírus gigantes no ambiente e em organismos hospedeiros.
“A cada novo genoma, aparecem genes completamente inéditos. Isso mostra que estamos lidando com um grupo cuja diversidade ainda é imensa”, afirma o professor. Essa coleção de genes, chamada de pangenoma, é crescente – ainda existem muitas espécies a serem descobertas. Para Del Bem, é um sinal claro: “Ainda não vimos tudo que esses vírus têm a oferecer”.
O artigo Integrative genomic analyses support the division of the extended Asfarviridae clade into multiple viral families está disponível neste link.
Matéria: Theo Schwan | Jornal da USP.